





Bild: Das Medaka-Retinal mit einer Mutation im ALG2-Gen zeigt im Gegensatz zu Wildtyp-Exemplaren einen allmählichen Verlust (lila Farben) von Stäbchenzellen (links). Diese Show… Show Mehr
Bildnachweis: Clara Baker
Ein seltener genetischer Defekt des sogenannten ALG2-Gens kann beim Menschen schwere Stoffwechselerkrankungen verursachen. Dies geschieht durch die fehlerhafte Bildung von Proteinen und Zuckermolekülen. Bis heute haben ihre Seltenheit und Komplexität die Untersuchung angeborener Glykosylierungsstörungen erschwert. Einem Forscherteam um Prof. Dr. Joachim Wittbrot und Dr. Thomas Thumberger vom Center for Organic Studies (COS) der Universität Heidelberg ist es nun endlich gelungen, die latente Mutation des ALG2-Gens in ein Fischmodell einzubringen und damit die Ursachen dieser komplexen Erkrankungen sollen auf molekularer Ebene untersucht werden.
Menschliche Zellen überleben durch die Aktivität von Millionen von Proteinen. Im ausgereiften Zustand müssen diese Proteine auf vielfältige Weise modifiziert werden, beispielsweise durch Zugabe von Zuckermolekülen – eine entscheidende Veränderung der richtigen Funktion. Fehler bei der Zuckerzugabe, auch Zuckerdekoration genannt, sind in sehr frühen Entwicklungsstadien oft tödlich. Wie Professor Wittbrot erklärt, führt ein genetischer Defekt in seltenen Fällen zu einem Mangel an Zuckerzusatz, der sich dann als angeborene Glykosylierungsstörung manifestiert. „Die Bindung des Proteins an die richtige Glykosylierung erfordert eine Reihe von Enzymen, die wie eine Uhr zusammenarbeiten“, sagt der Forscher. Eine besonders wichtige Aufgabe übernimmt dabei das ALG2-Gen. Es kodiert ein Enzym, das für die korrekte Verzweigung der Zuckerkette notwendig ist. Ist dieser Prozess gestört, erscheinen die Patienten bei der Geburt unbeeinflusst, entwickeln aber in der frühen Kindheit Probleme in verschiedenen Organen wie Augen, Gehirn und Muskeln.
Das Team um Professor Whitbrot und Dr. Thumberger nutzte die CRISPR/Cas9-Gen-Editing-Schere, um die ALG2-Mutation in ein Fischmodell, den japanischen Reisfisch oder Medaka, einzuführen. „Fische sind besonders gute Modelle für diese Erkrankungen, weil sie sich außerhalb der Mutter entwickeln und sich daher sehr gut für die Untersuchung früher embryonaler Defekte eignen“, erklärt Dr. Thumberger. Zudem lässt sich das Genom des japanischen Reisfisches effizient und präzise bearbeiten. „Unser Fisch ist sozusagen ein genetischer Zwilling, sodass die Wirkung einzelner Veränderungen im Vergleich zu nicht-transgenen Fischen direkt bestimmt werden kann.“
Obwohl die evolutionäre Distanz zwischen Mensch und Fisch groß ist, haben Forscher viele der gleichen Symptome im Fischmodell berichtet, die bei ALG2-Patienten beobachtet wurden, einschließlich bestimmter neurologischer Defekte. Sie waren überrascht von den Ergebnissen der Analyse des gesamten Medaka-Organismus, die das gesamte Spektrum verschiedener Zelltypen berücksichtigte. „Obwohl alle Fischzellen die gleiche reduzierte ALG2-Aktivität zeigten, waren einige Zelltypen stärker betroffen als andere“, sagt Professor Wittbrot. In der Netzhaut des Fisches waren die Zapfenzellen, die für die Farbwahrnehmung benötigt werden, nicht betroffen, aber es gab einen allmählichen Verlust der Stäbchenzellen, die zum Sehen bei schwachem Licht erforderlich waren, was den Fisch mit Nachtblindheit zurückließ. Die Forscher hoffen nun, die Proteine zu identifizieren, die den Zelltod von Bacillus aufgrund einer verminderten Zuckerbindung verursachen.
„Unsere Studien im Medaka-Fischmodell haben gezeigt, dass durch die Bereitstellung voll funktionsfähiger ALG2-mRNA – der Bauplan für die korrekte ALG2-Enzymproduktion – alle Symptome verhindert werden können. Wir konnten den Gendefekt im Fischmodell effektiv umkehren. Das heißt, wir können“ analysieren nun systematisch einzelne Funktionsbereiche für das ALG2-Enzym.Uns interessiert insbesondere die zelltypspezifische Antwort im Gesamtorganismus“, betont Joachim Wittbrot. Darauf aufbauend will das Heidelberger Forscherteam die molekularen Mechanismen und Ursachen der Entstehung solch komplexer Stoffwechselerkrankungen beim Menschen untersuchen.
###
An der Studie beteiligten sich neben dem Heidelberger Team am COS auch Forscher des Zentrums für Molekularbiologie (ZMBH) der Universität Heidelberg, des Universitätsklinikums Heidelberg und des Max-Planck-Instituts für komplexe technische Systemdynamik in Magdeburg. Die Forschungsarbeiten wurden von der Deutschen Forschungsgemeinschaft gefördert. Die Ergebnisse der Studie wurden in der Zeitschrift veröffentlichtEntwicklung„.
Haftungsausschluss: AAAS und EurekAlert! Nicht verantwortlich für die Richtigkeit der an EurekAlert gesendeten Newsletter! Durch die beitragenden Institutionen oder für die Nutzung von Informationen über das EurekAlert-System.

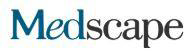


More Stories
Die deutsche Luft- und Raumfahrtindustrie steht vor dem Ende des Typhoon-Programms
Airbus erhält 2,1 Milliarden Euro für den Aufbau eines deutschen militärischen Kommunikationssystems
RFA, ATMOS und Yuri kündigten den Mikrogravitationsdienst „Eva“ an